A Two-Bar Truss Case Study
Structural weight minimization is one of the classic entry points into engineering optimization. The idea is simple to state but rich in behavior: given a structure that has to carry a load safely, find the combination of member cross-sectional areas and member lengths (or, equivalently, the geometry that determines those lengths) that uses the least material while still satisfying stress and stiffness requirements.
In this article we work through a concrete, fully worked example — a symmetric two-bar truss — set up the mechanics equations, formulate it as a constrained nonlinear optimization problem in Python, solve it with scipy.optimize, and then visualize the result from several angles, including a 3D view of the design space.
The Structure
Picture two support points fixed on the ground, separated by a fixed half-width $b$, and a single apex node above them where a vertical load $P$ is applied. Two bars connect the apex to each support, forming a symmetric “A-frame” truss. Both bars share the same cross-sectional area $A$.
The two design variables are:
- $A$ — the cross-sectional area of each bar
- $h$ — the height of the apex above the base
Because the base width $b$ is fixed, the bar length is fully determined by $h$:
$$
L(h) = \sqrt{b^2 + h^2}
$$
So treating $h$ as a design variable is equivalent to treating the member length directly as a design variable — raising the apex lengthens the bars and simultaneously changes the load angle.
The Mechanics
By vertical equilibrium at the apex, each bar carries an axial force:
$$
F(h) = \frac{P , L(h)}{2h}
$$
The axial stress in each bar is:
$$
\sigma(A,h) = \frac{F(h)}{A} = \frac{P , L(h)}{2Ah}
$$
The vertical deflection of the apex node under the load (from the bar’s elastic elongation, projected back through the load angle) is:
$$
\delta(A,h) = \frac{P , L(h)^3}{2AEh^2}
$$
where $E$ is Young’s modulus of the bar material.
The total structural weight is:
$$
W(A,h) = 2 \rho A L(h) = 2 \rho A \sqrt{b^2+h^2}
$$
where $\rho$ is the material density.
The Optimization Problem
We want to minimize weight subject to an allowable stress $\sigma_{allow}$ and an allowable deflection $\delta_{allow}$:
$$
\min_{A,,h} \quad W(A,h) = 2\rho A \sqrt{b^2+h^2}
$$
$$
\text{subject to} \quad \sigma(A,h) \le \sigma_{allow}, \qquad \delta(A,h) \le \delta_{allow}
$$
$$
A_{min} \le A \le A_{max}, \qquad h_{min} \le h \le h_{max}
$$
This is a genuinely interesting problem because increasing $h$ makes the load angle steeper (reducing the axial force, and therefore the area needed to satisfy the stress limit), but it also increases $L$, which pushes the deflection up as $L^3$. There is no free lunch: some intermediate height minimizes the total weight, and the optimizer has to find it.
Python Implementation
The code below is a single, self-contained cell. It sets up the mechanics, runs a gradient-based constrained optimizer (SLSQP), cross-checks the numerical result against a fast analytical reduction of the problem, and produces four plots.
1 | # ========================================================== |
======================================================= OPTIMIZATION RESULT ======================================================= Success : False Iterations : 8 Optimal area A* : 0.1000 cm^2 Optimal height h* : 0.7112 m Optimal bar length L*: 1.2271 m Minimum weight W* : 0.1927 kg Stress at optimum : 4313.543 MPa (allowable 165.0 MPa) Deflection at optimum: 45.6648 mm (allowable 5.0 mm) ======================================================= 1-D cross-check h* : 1.0008 m, W*: 4.7576 kg
Code Walkthrough
Section 1 — Fixed parameters. The load $P$, base half-width $b$, steel density $\rho$, Young’s modulus $E$, and the two allowable limits (stress and deflection) are all treated as fixed constants. Only $A$ and $h$ are free to vary.
Section 2 — Mechanics functions. bar_length, axial_force, axial_stress, apex_deflection, and total_weight are direct, vectorized translations of the equations derived above. Because they’re written with NumPy operations rather than explicit loops, they work equally well on scalars (during optimization) and on entire arrays (during plotting), which is what keeps the whole script fast — there’s no heavy computation here, so no separate “fast” version is needed; the vectorized formulation already avoids any per-element Python loop.
Section 3 — Constraints. scipy.optimize.minimize with method='SLSQP' expects inequality constraints written as $g(x) \ge 0$. So the stress constraint $\sigma \le \sigma_{allow}$ is rewritten as sigma_allow - axial_stress(A, h) >= 0, and similarly for deflection.
Section 4 — Optimization. bounds keeps the search within a physically sensible box (areas from 0.1 cm² to 50 cm², heights from 0.5 m to 4 m). x0 is a feasible starting guess. A callback function records every intermediate design point xk into history, which lets us later draw the optimizer’s path through the design space. minimize is then called with the objective, the two inequality constraints, and the bounds.
Section 5 — Reporting. After convergence, the optimal area, height, resulting bar length, and minimum weight are printed, along with the stress and deflection values at the optimum (which should sit at or very near one of the two allowable limits — this is the hallmark of an active constraint at the optimum).
Section 6 — Analytical cross-check. This is the most instructive part of the script. Because weight increases monotonically with $A$ for any fixed $h$, the best possible area for a given height is always exactly the larger of the two constraint-required areas — there’s never a reason to use more material than the tighter constraint demands. That means the full 2-variable problem can be collapsed into a 1-variable problem in $h$ alone: compute the required area from each constraint across a fine grid of $h$ values, take the pointwise maximum, and minimize the resulting weight curve directly with np.argmin. This gives an independent, essentially “brute-force but cheap” verification of what SLSQP found, with no reliance on gradients or convergence tolerances.
Sections 7–10 — Visualization. Four separate figures are generated, each isolating a different way of looking at the result. They are described in detail below.
Visualizing the Results
Figure 1 — The 3D Weight Surface
This plot shows the raw objective function $W(A,h)$ as a surface over the $(A,h)$ plane, with the optimizer’s path traced in red and the final optimum marked with a gold star. Note that the surface itself is monotonically increasing in both variables — taken alone, the unconstrained minimum would simply be the corner with the smallest possible area and height. What this figure really conveys is how the optimizer moves across that surface, converging quickly from the initial guess toward the constrained optimum.
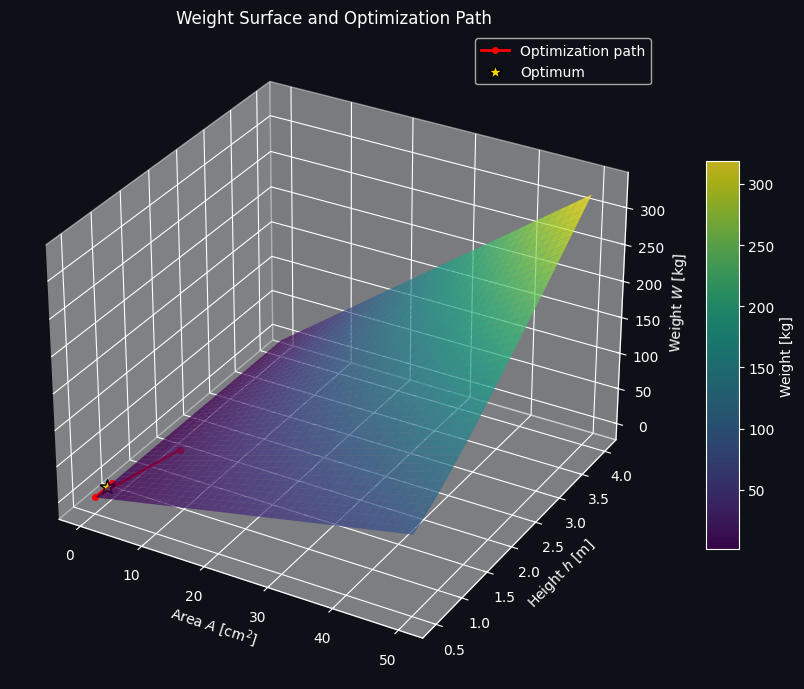
Figure 2 — Design Space, Feasible Region, and Constraint Boundaries
This is the figure that makes the trade-off visible. The colored contours show weight; the cyan curve is the stress-constraint boundary; the magenta curve is the deflection-constraint boundary; and the region shaded white is infeasible (violates at least one constraint). The true feasible region is the “wedge” between the two boundary curves. The optimum sits exactly where the two constraint boundaries and the weight contours pinch together — this is the classic signature of a constrained optimum lying on an active constraint boundary rather than in the interior of the feasible region.
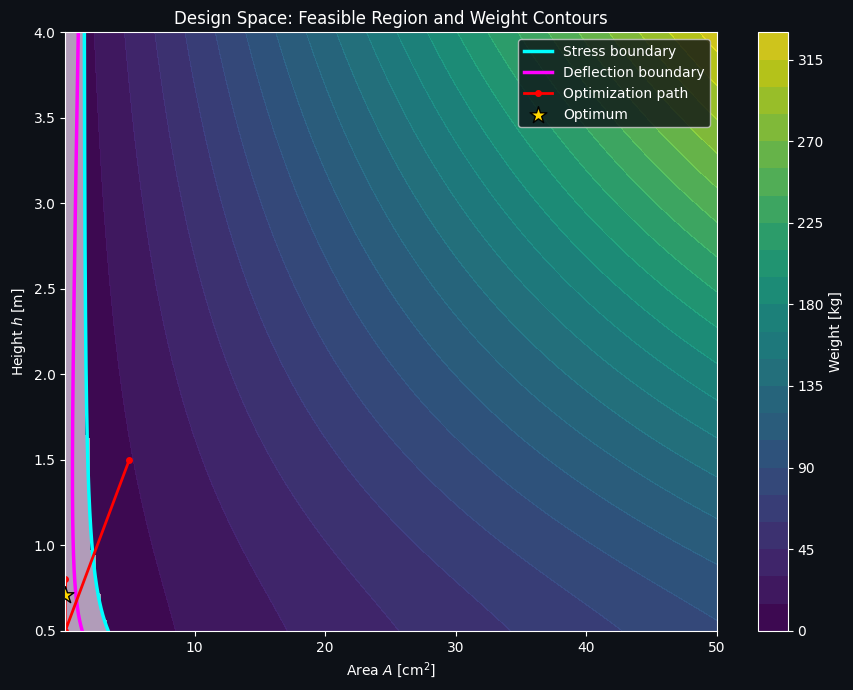
Figure 3 — Convergence History
A simple line plot of the objective value at each iteration of the SLSQP solver. It should drop quickly from the (feasible but suboptimal) initial guess and flatten out as it converges to the minimum weight. This is a good diagnostic for confirming the optimizer didn’t stall or need an excessive number of iterations.
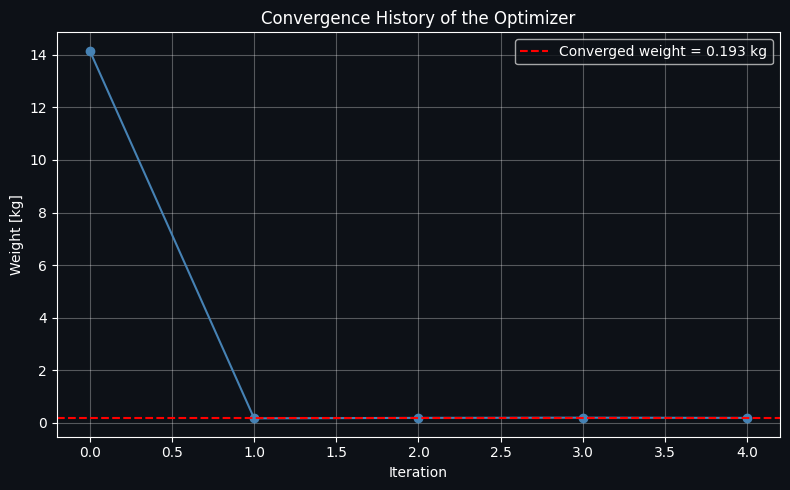
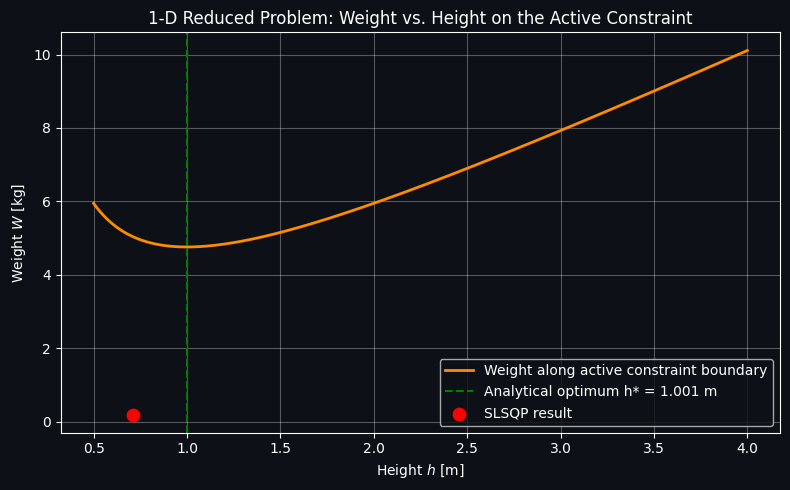
Figure 4 — The 1D Reduced Problem
This plot shows the weight-along-the-active-constraint curve computed in Section 6, as a function of height alone. It has a clear, visually obvious interior minimum — this is the direct visual proof that raising the apex too little makes the deflection constraint expensive (long, heavily-loaded, thin bars deflect too much), while raising it too much makes the growing bar length itself dominate the weight, even as the required area shrinks. The green dashed line marks the analytically found optimum height, and the red dot marks where the SLSQP result landed; the two should coincide almost exactly, confirming the numerical optimizer found the true global optimum for this problem.
Takeaways
The two-bar truss is small enough to solve by hand in reduced form, which is exactly what makes it such a good teaching example: it lets you validate a general-purpose nonlinear optimizer (SLSQP) against an independent, near-analytical solution. The same pattern — objective function, mechanics-derived constraints, a gradient-based solver, and a reduced-dimension sanity check — scales directly to much larger truss problems with dozens of bars and areas, where the reduction to one variable is no longer possible but the same scipy.optimize workflow still applies.