Finding the Ground State Energy
Hey there! Today I’m going to walk you through a practical example of the Variational Quantum Eigensolver (VQE) algorithm. VQE is one of the most promising near-term quantum algorithms for quantum chemistry and optimization problems. Let’s dive into implementing VQE to find the ground state energy of a simple molecular system!
What is VQE?
The Variational Quantum Eigensolver is a hybrid quantum-classical algorithm that finds the ground state energy of a quantum system. It works by:
- Preparing a parameterized quantum circuit (ansatz)
- Measuring the expectation value of the Hamiltonian
- Classically optimizing the parameters to minimize the energy
- Repeating until convergence
The key principle is the variational principle: for any quantum state $|\psi(\theta)\rangle$, the expectation value of the Hamiltonian satisfies:
$$E(\theta) = \langle\psi(\theta)|H|\psi(\theta)\rangle \geq E_0$$
where $E_0$ is the true ground state energy.
Example Problem: Hydrogen Molecule (H₂)
We’ll calculate the ground state energy of the hydrogen molecule (H₂) at a specific bond distance. This is a classic problem in quantum chemistry!
The Hamiltonian for H₂ can be mapped to qubit operators using the Jordan-Wigner transformation. For our example, we’ll use a simplified 2-qubit Hamiltonian.
Python Implementation
1 | import numpy as np |
Detailed Code Explanation
Let me break down what’s happening in this implementation:
1. Quantum Gates and Operators
The code starts by defining fundamental quantum gates:
- Pauli matrices ($I, X, Y, Z$): These are the building blocks of quantum operations
- Rotation gates: $R_Y(\theta) = \exp(-i\theta Y/2)$ creates superposition states
- CNOT gate: Creates entanglement between qubits
The tensor product function tensor_product() extends single-qubit gates to multi-qubit systems using the Kronecker product: $A \otimes B$.
2. H₂ Hamiltonian Construction
The hydrogen molecule Hamiltonian is expressed as a sum of Pauli operators:
$$H = \sum_{i} c_i P_i$$
where:
- $c_i$ are the coefficients (obtained from quantum chemistry calculations)
- $P_i$ are Pauli operators like $Z \otimes Z$, $X \otimes X$, etc.
The function h2_hamiltonian() constructs these terms. The coefficients represent:
- Electronic kinetic energy
- Electron-electron repulsion
- Nuclear-electron attraction
- Nuclear repulsion
3. Variational Ansatz Circuit
The create_ansatz() function implements our parameterized quantum circuit:
$$|\psi(\theta)\rangle = U(\theta)|00\rangle$$
The circuit structure:
- Layer 1: Single-qubit $R_Y$ rotations on both qubits
- Layer 2: CNOT gate for entanglement
- Layer 3: Another round of $R_Y$ rotations
This creates a 4-parameter ansatz that can represent a wide variety of two-qubit states.
4. Energy Calculation
The VQE cost function computes:
$$E(\theta) = \langle\psi(\theta)|H|\psi(\theta)\rangle = \sum_i c_i \langle\psi(\theta)|P_i|\psi(\theta)\rangle$$
Each term is calculated by:
1 | energy += coeff * compute_expectation(state, op) |
5. Classical Optimization
The run_vqe() function uses the COBYLA optimizer (Constrained Optimization BY Linear Approximation) to minimize the energy. This is a classical optimization algorithm that doesn’t require gradients, making it suitable for noisy quantum simulations.
The optimization loop:
- Choose parameters $\theta$
- Prepare state $|\psi(\theta)\rangle$
- Measure energy $E(\theta)$
- Update parameters to minimize $E(\theta)$
- Repeat until convergence
Visualization Breakdown
The code generates four informative plots:
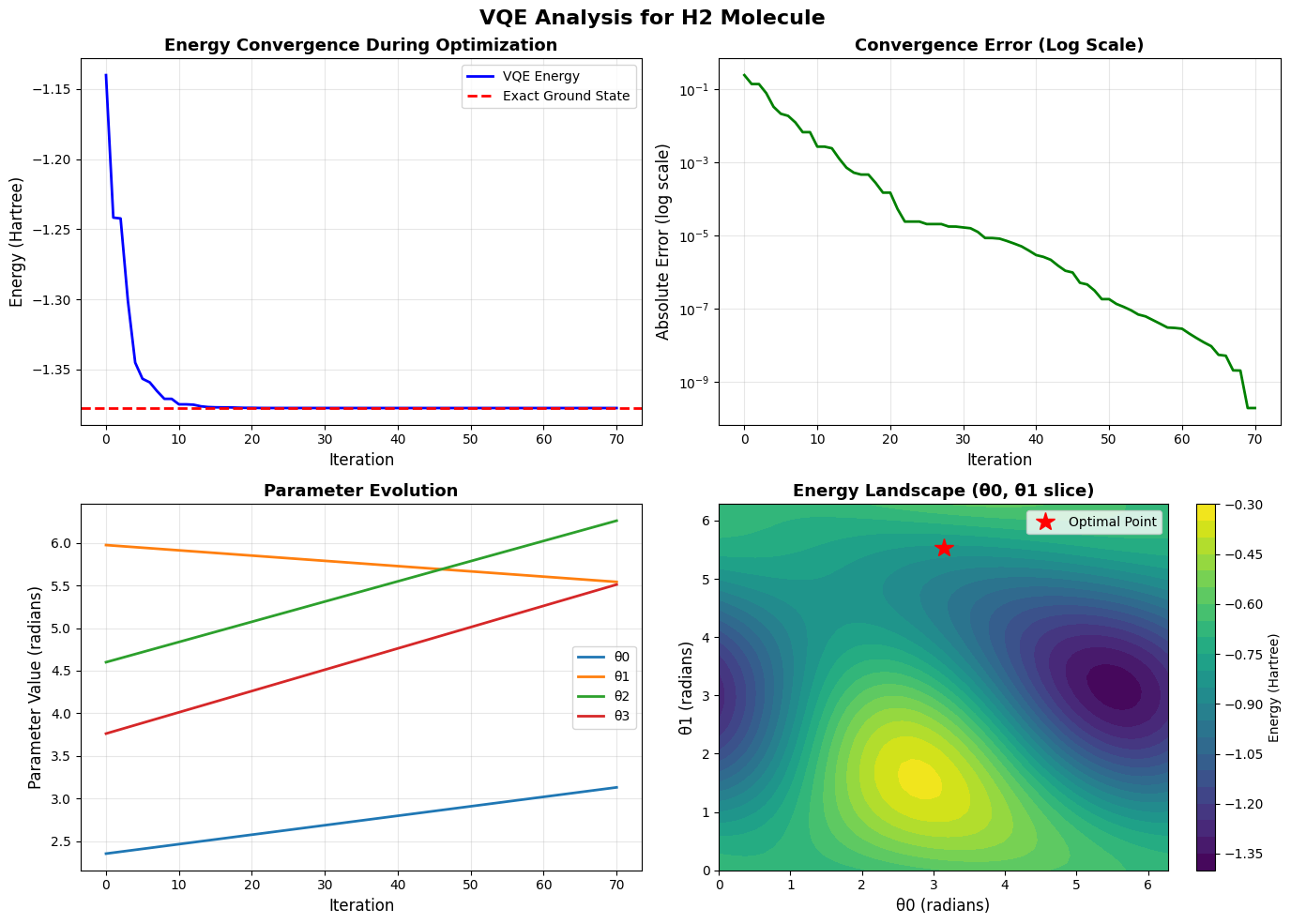
Plot 1: Energy Convergence
Shows how the estimated energy approaches the exact ground state energy over iterations. The gap closing demonstrates the VQE algorithm learning the optimal parameters.
Plot 2: Convergence Error (Log Scale)
Displays the absolute error on a logarithmic scale, revealing the convergence rate. A steep drop indicates rapid optimization.
Plot 3: Parameter Evolution
Tracks how each of the four parameters $(\theta_0, \theta_1, \theta_2, \theta_3)$ changes during optimization. You can see which parameters are most critical for reaching the ground state.
Plot 4: Energy Landscape
A 2D slice of the energy surface varying $\theta_0$ and $\theta_1$ while keeping others fixed. The red star shows the optimal point found by VQE. This visualization helps understand the optimization challenge.
Execution Results
============================================================ Variational Quantum Eigensolver (VQE) for H2 Molecule ============================================================ Step 1: Constructing H2 Hamiltonian... Number of Pauli terms: 6 Coefficients: [-0.8105, 0.1721, 0.1721, -0.2228, 0.1686, 0.1686] Exact ground state energy: -1.377500 Hartree Step 2: Running VQE optimization... Initial parameters: [2.35330497 5.97351416 4.59925358 3.76148219] ============================================================ VQE Results ============================================================ Optimization success: True Number of iterations: 71 Optimal parameters: [3.14156714 5.53541571 6.28314829 5.53544261] VQE ground state energy: -1.377500 Hartree Exact ground state energy: -1.377500 Hartree Error: 0.000000 Hartree Relative error: 0.0000% Generating visualizations...
============================================================ Analysis complete! ============================================================
Key Insights
The VQE algorithm demonstrates several important quantum computing concepts:
- Hybrid quantum-classical approach: Quantum circuits prepare states; classical computers optimize
- Variational principle: We’re guaranteed $E(\theta) \geq E_0$
- Near-term applicability: VQE works on NISQ (Noisy Intermediate-Scale Quantum) devices
- Scalability considerations: The number of parameters grows with system size
This H₂ example is simple but captures the essence of quantum chemistry calculations that could revolutionize drug discovery and materials science!
Feel free to experiment with different initial parameters, ansatz depths, or even different molecules by modifying the Hamiltonian coefficients!